Rapid evolution of an adaptive multicellular morphology of Candida auris during systemic infection - Nature.com
Abstract
Candida auris has become a serious threat to public health. The mechanisms of how this fungal pathogen adapts to the mammalian host are poorly understood. Here we report the rapid evolution of an adaptive C. auris multicellular aggregative morphology in the murine host during systemic infection. C. auris aggregative cells accumulate in the brain and exhibit obvious advantages over the single-celled yeast-form cells during systemic infection. Genetic mutations, specifically de novo point mutations in genes associated with cell division or budding processes, underlie the rapid evolution of this aggregative phenotype. Most mutated C. auris genes are associated with the regulation of cell wall integrity, cytokinesis, cytoskeletal properties, and cellular polarization. Moreover, the multicellular aggregates are notably more recalcitrant to the host antimicrobial peptides LL-37 and PACAP relative to the single-celled yeast-form cells. Overall, to survive in the host, C. auris can rapidly evolve a multicellular aggregative morphology via genetic mutations.
Similar content being viewed by others

Rapid proliferation due to better metabolic adaptation results in full virulence of a filament-deficient Candida albicans strain
The hyphal-specific toxin candidalysin promotes fungal gut commensalism
Forward and reverse genetic dissection of morphogenesis identifies filament-competent Candida auris strains
Introduction
The emerging human fungal pathogen Candida auris was first described in 2009 in Japan and is becoming a serious global health threat1,2,3. Due to the notable increase in the incidence of C. auris infections worldwide, the rapid transmission and outbreak potential of C. auris, its multidrug resistance properties, and its persistence in the environment, the Centers for Disease Control and Prevention (CDC; https://www.cdc.gov) has issued several clinical alerts to healthcare facilities to be on the lookout for C. auris infections in patients and the World Health Organization (WHO; http://www.who.int) has added C. auris to the 2022 fungal priority pathogens list4. Like other pathogenic Candida species such as Candida albicans and Candida tropicalis, C. auris is able to colonize the ears, wounds, respiratory tracts, urinary tracts, and skin of immunocompetent individuals, and can also cause serious bloodstream and/or invasive infections largely in immunocompromised patients2,3.
Given its recent emergence and rapid spread worldwide, understanding the mechanisms of how C. auris rapidly evolved from a benign environmental fungus to a successful fungal pathogen is of utmost importance to controlling and preventing outbreaks of C. auris and other emerging fungal pathogens in the future. Morphological plasticity is a general and critical strategy used by pathogenic bacteria and fungi to adapt to changing environmental conditions and survive in the host5,6,7. For example, C. albicans can switch between multiple morphological phenotypes in response to in vivo and in vitro environmental changes8. White-opaque switching and yeast-filament transitioning are among the most well-studied morphological transitions investigated in C. albicans over the past two decades8,9,10. Different morphological cell types of C. albicans often have distinct characteristics in term of host invasion, stress resistance, tissue tropism, and susceptibility to antifungal drugs and the host immune system8,9,10. For example, in C. albicans, multicellular filaments are more invasive than yeast-form cells, while white cells are more virulent than opaque cells in mouse models of systemic infections10.
Numerous environmental factors and genetic regulators and pathways are involved in the control of morphological transitions in C. albicans8,10,11,12. For example, increased ambient temperatures and CO2 levels, serum, neutral or basic pH, and N-acetylglucosamine promote the growth of filaments, while decreased ambient temperatures, acidic pH, and rich nutrient conditions induce the transition from the filamentous to the yeast form11. The cAMP/PKA signaling and the Ste11-Hst7-Cek1/2 MAPK pathways function as major regulators in controlling filamentation in C. albicans11,13,14. Activation of these two conserved pathways leads to changes in expression of downstream transcription factors and ultimately the development of filaments. Moreover, genes involved in the regulation of cell wall integrity, formation of septin rings, cytokinesis, and cytoskeletal polarization also often play important roles in mediating morphological transitions in C. albicans11,15,16. Environmental cues induce morphological transitions in pathogenic Candida species largely through the regulation of the aforementioned signaling pathways by epigenetic or non-genetic changes11,12,15,16. Recent studies have additionally shown that genetic mutations are also involved in shaping the virulence or commensal characteristics of certain Candida species17,18,19.
In response to host environmental stresses, pathogenic Candida species often undergo microevolution driven by genetic mutations to adapt to the host during commensal colonization or infection18,19,20,21,22,23. For example, serial passage of C. albicans through the mouse gut induced the adaptive evolution of a low-virulence phenotype with an enhance ability to colonize the mammalian gut18. Passaging C. albicans in this way led to mutations in several genes required for C. albicans filamentous growth, including FLO8 and EFG1. In another example, continuous coincubation of Candida glabrata with host murine macrophages led to the formation of a pseudohyphal-like morphology and a mutation in the chitin synthase-encoding CHS2 gene22. This evolved pseudohyphal-like form allowed C. glabrata to rapidly escape from macrophages and the pseudohyphal-like cells displayed increased virulence during host infection. The soil fungal pathogen Cryptococcus neoformans is also capable of undergoing microevolution to survive in the host. Coincubation of C. neoformans with its environmental amoeba predator has been shown to induce a pseudohyphal form of C. neoformans caused by DNA mutations in genes encoding components of the Regulation of Ace2 Morphogenesis (RAM) pathway23. Taken together, numerous pathogenic fungi undergo modifications of their genomic sequences to enhance their abilities to adapt to changing environmental conditions that occur during host colonization and infection. These small-scale genetic changes may be efficient drivers of microevolution in these species.
Like C. albicans, C. auris also has several cellular morphologies including yeast and filamentous forms2. It has been demonstrated that morphological transitions are a general feature of clinical C. auris strains of different genetic clades24. A number of transcriptional and cell wall regulators are involved in the regulation of filamentous growth and aggregation in C. auris25. Genotoxic stress and inactivation of the molecular chaperone Hsp90 also promote the growth of filaments in C. auris26,27, indicating that its ability to undergo morphological transitions has conserved regulatory features. One study demonstrated that DINOR, a long non-coding RNA, plays a critical role in the control of filamentation and virulence in C. auris28. Deletion of DINOR resulted in DNA damage and filamentous growth, suggesting that genetic alterations could be involved in the regulation of morphogenesis in C. auris. Interestingly, clinical isolates of C. auris rarely undergo filamentation, although a subset of C. auris cells can become filament-competent (acquiring the ability to develop filaments) after passage through the mammalian host29. The mechanism underlying this host-induced filamentation in C. auris remains to be investigated. Since the filamentous and filament-competent cells exhibit cellular memory of their cell state, we suspect that genetic or epigenetic changes could be involved in this regulation2.
Another striking feature of C. auris is its ability to form multicellular aggregates25,30,31,32. Numerous clinical isolates of C. auris can form aggregative cells that differ from single-celled yeast-form cells in a number of biological aspects including colony and cellular morphologies, biofilm-forming abilities, susceptibilities to antifungal drugs, susceptibilities to immune cells, and virulence24,25,30,31,32,33. The aggregative form could be induced under both in vivo and in vitro conditions24,25,30,31,32. For example, exposure to the antifungal drug caspofungin, an inhibitor of β−1,3-glucan synthase, caused not only modifications of the cell wall but also led to morphological changes and the formation of aggregative cells in C. auris34. Forgacs et al. 35 observed the presence of large aggregates of C. auris cells in the heart, kidney, and liver tissues in a neutropenic mouse model, suggesting that numerous clinical isolates are capable of forming aggregates in the host35. We and others recently found that there are two types of aggregative morphologies in C. auris30,36,37. One type is formed due to a defect in cell division, while the other type is thought to be the result of the overexpression of genes associated with adhesion. These two aggregative morphologies of C. auris differ in their biofilm forming abilities and virulence30. The host environmental inducers, biological bases, and regulatory mechanisms of the former aggregative phenotype, however, remain unknown.
In this study, we report that a subset of C. auris cells can rapidly evolve from the single-celled yeast form to a multicellular aggregative morphology during systemic host infection in a mouse model. Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) analyses indicate that formation of this aggregative phenotype was caused by cell division or budding defects. Genetic mutations in genes associated with the regulation of cell wall integrity, cytokinesis, cytoskeletal properties, and cellular polarization were responsible for the rapid evolution of this multicellular aggregative phenotype. Compared to the yeast-form morphology, these C. auris aggregative cells exhibited a general increased ability to survive in the host and an increased capacity to colonize the brain. Moreover, aggregative cells were more resistant to phagocytosis by host macrophages and host-derived antimicrobial peptides. This rapidly evolved aggregative morphology likely represents an adaptive strategy of C. auris in response to the host environment and attack by the host immune system.
Results
Aggregate formation in C. auris can be induced during systemic infection in the host
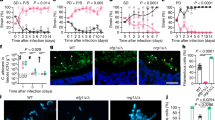
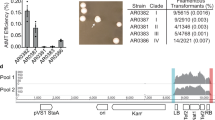
A unique characteristic of C. auris is that some clinical isolates exhibit a multicellular aggregative phenotype31,32,33,38. As shown in Fig. 1 and in previous studies30,31,32,33, this multicellular morphology is associated with defects in cell division. The C. auris mother and daughter cells within the multicellular aggregates remain attached caused by an inability to release daughter cells after budding. In our previous studies, we observed that rough or wrinkled colonies of C. auris occasionally contained filamentous or aggregative cells when C. auris was recovered from mouse tissues during systemic infection assays24,29. To reveal whether the mammalian host is able to induce the formation of C. auris aggregates, we revisited these mouse systemic infection experiments in more detail (Fig. 2a). C. auris yeast-form cells of 11 clinical strains spanning the four major C. auris genetic clades were injected into the mice via the tail vein (Table S1). After 3 days of infection, we recovered fungal cells from the brain, liver, spleen, lung, and kidney and replated them onto YPD medium containing the red dye phloxine B. This dye was used in the screening assay because it largely stains colonies containing aggregative cells of C. auris pink or red and facilitated our abilities to identify evolved mutant strains. The average frequency of aggregative mutant strains from brain tissue was 0.011%, whereas the average frequency from the liver, spleen, lung, and kidney tissues were 0.0019%-0.0086%. Of note, the fungal burden in the blood was extremely low and we did not obtain aggregative mutants from the bloodstream. As shown in Fig. 2b, in total, we identified 96 rough or pink/red colonies that contained aggregative cells of C. auris from the mice infected with 8 strains (8/11, 72.72%). Of them, the majority (54/96, 56.25%) were isolated from the brain, implying that aggregative cells have a tissue tropism for the mouse brain during systemic infections. The morphologies of the aggregative cells at the cellular level were examined using differential interference contrast (DIC) microscopy, SEM, and TEM assays. Several representative cellular morphologies are shown in Figs. 2c and S1. Our results indicate that the mammalian host provides a selective advantage for cells capable of generating the multicellular aggregative morphology during systemic infections.

Strain used: AR0386 and AR0386A. C. auris cells were plated onto YPD plates at 30 °C for 4 days. DIC differential interference contrast, SEM scanning electron microscopy, TEM transmission electron microscopy. Red arrows indicate intercellular septa. Scale bar for DIC, 10 μm; for SEM and TEM, 2 μm.

a Schematic of aggregative colony acquisition from a mouse systemic infection system. Six-week-old mice were injected with the yeast-form cells of C. auris via the tail vein. Fungal cells were recovered from the brain, liver, spleen, lung, and kidney tissues after 3 days post infection (dpi) and plated onto YPD medium plates containing the red dye phloxine B. Pink, rough or wrinkled colonies containing aggregative cells were subject to microscopy assays and whole genome sequencing (WGS). b Number of evolved aggregative C. auris strains isolated from different mouse organs. In total, 113 aggregative mutant strains were obtained. Of them, 96 strains (black rectangles) were isolated from the brain, liver, spleen, lung, and kidney tissues of 28 mice and 17 strains (gray rectangle) were isolated from the brain tissue of 12 mice. Detailed strain information is presented in Tables S1, S2, and Dataset S1. c Colony and cellular morphologies of the WT (BJCA001) and four representative aggregative strains evolved during mouse systemic infections. CHS1 p.Q826* (FDAG4), BNI1 p.206 fs (FDAG30), LRG1 p.K427* (FDAG9), and CAS4 p.1288 fs (FDAG1) represent the four evolved strains with mutations in CHS1, BNI1, LRG1, or CAS4, respectively. C. auris cells were plated onto YPD plates supplemented with phloxine B at 30 °C for 4 days. *, nonsense mutations; fs, frameshift mutations. Red arrows indicate intercellular septa. Scale bar for colony, 5 mm; for DIC morphology, 10 μm; for SEM and TEM morphologies, 2 μm.
To exclude the possibility of potential bias being introduced in our mutation screen due to the use of the red dye phloxine B, we performed comparative experiments using media with and without phloxine B. Brain tissues from 12 C. auris infected mice were broken up and equally plated onto YPD plates with and without phloxine B. We identified 11 evolved aggregative mutant strains from the medium containing phloxine B and 6 mutant strains from the medium lacking phloxine B (Table S2 and Dataset S1). Therefore, the addition of phloxine B facilitated our abilities to identify evolved mutant strains and did not introduce bias in our mutation screen.
Genetic mutations underlie the rapid evolution of the C. auris multicellular aggregative morphology
We observed that C. auris cells within all 113 identified colonies with a rough or red appearance exhibited heritability for the aggregative phenotype under in vitro culture conditions, suggesting that genetic or epigenetic alterations could be at play in the regulation of this multicellular aggregative morphology. To uncover the underlying regulatory mechanism, we performed next generation whole-genome sequencing (WGS) with all evolved aggregative strains. Single-nucleotide polymorphism (SNP) and copy number variation (CNV) analyses were performed, and the detailed results are presented in Dataset S1. To our surprise, all aggregative strains isolated from the mouse host carried one or more mutations or copy number losses in the open reading frame (ORF) regions of a set of genes associated with cell division and/or budding. In total, 48 distinct mutations were identified, which involved 31 independent genes (Fig. 3a and Dataset S1). We observed missense, nonsense, frameshift, and gene loss mutations within these regions (Dataset S1). Functional category analysis indicated that the major of mutated C. auris genes in these regions were enriched for genes encoding proteins involved in the regulation of cell wall integrity, cytokinesis, cytoskeletal properties, and/or cellular polarity (Figs. 3 and S2). Notably, mutations within the chitin synthase-encoding gene CHS1 were found in 39 evolved aggregative C. auris strains and included four distinct types of mutations (Dataset S1). Chs1 is required for primary septum synthesis and cell wall integrity in C. albicans39. Similar to its C. albicans counterpart, CHS1 in C. auris encodes a class II chitin synthase. Mutation of CHS1 led to a potential aberrant septa in C. auris cells (Fig. 2c). Other notable mutated C. auris genes included KIN3 (14 isolates), ACE2 (8 isolates), CAS4 (6 isolates), LRG1 (5 isolates), APM1 (4 isolates), BNI1 (4 isolates), KIC1 (3 isolates), and LAA1 (3 isolates), which are all involved in cytokinesis, cytoskeletal properties, and/or cellular polarity processes. A summary of the associated signaling pathways and cellular processes for the identified mutated genes is presented in Fig. 3b.

These genes were independently identified from different evolved aggregative mutant strains. a Number of isolates for each gene mutation. All aggregative mutant strains were recovered from mouse organs initially infected with yeast-form C. auris cells (as shown in Fig. 2). The mutated loci were identified by comparing the genomic sequences of the mutant strains with those of the yeast-form parent strain. b Major biological processes or signaling pathways of the mutated genes (highlighted in red) of the evolved aggregative strains. Cell wall integrity pathway: 14 mutations were identified, including mutations in genes encoding chitin synthase Chs1, GTPase regulator Lrg1, MAPK proteins Dig2, and several cell wall proteins. Cytokinesis: 7 mutations were identified, including mutations in LRG1 and genes encoding the formin protein Bni1, F-BAR protein Hof1, and IQGAP protein. Actin cable and cytoskeleton: 8 mutations were identified, including mutations in genes encoding components of the actin cytoskeleton-regulatory complex Pan1, clathrin and adaptor complex (Apl2, Apl4, Apm1), and the Arp2/3 complex (Arc19). Cellular polarity: 6 mutations were identified, including mutations in LRG1, BNI1, and genes encoding kinases Kin3 and Hsl1 associated with septin formation. RAM pathway: 13 mutations were identified, including mutations in ACE2, CBK1, MOB2, HYM1, KIC1, and CAS4. c Schematic model indicates the cellular functions of the identified mutated genes. Most mutated genes are involved in the regulation of cell budding and/or cell division. The blue strip indicates the septum; red lines indicate actin cables involved in exocytosis; red circles indicate actin patches involved in endocytosis; orange circles indicate septin double rings in endocytosis; the green solid circle indicates protein Bni1; and the peripheral shadow layer indicates the cell wall. Mutated genes identified in this study are highlighted in red. The mutated genes/pathways identified in this study are interconnected and several genes are involved in the regulation of multiple processes. Note that the biological processes or signaling pathways of the C. auris mutated genes shown were predicted based on homology to their S. cerevisiae or C. albicans counterparts (b) and (c).
We note that several of the mutated C. auris genes (CBK1, MOB2, HYM1, KIC1, ACE2, and CAS4) identified in the C. auris evolved strains (Fig. 3) are related to fungal genes that are members of the well-conserved Regulation of Ace2 Morphogenesis (RAM) pathway, which regulates cellular polarity, cell separation, and filamentation in C. glabrata, C. albicans, and Saccharomyces cerevisiae40,41,42,43. The Ace2 transcription factor regulates chitinase (Cht1) that is required for septal plate dissolution. Consistently, Santana and O'Meara demonstrated that inactivation of Ace2 caused a cell separation defect in C. auris25. Tian et al. 44 recently isolated two aggregative C. auris clinical strains carrying ACE2 mutations44. Given their conserved roles in cell growth or the cell cycle in other fungi, it is reasonable that mutations in genes associated with these pathways could lead to defects in budding and/or cell division in C. auris.
Four genes (LRG1, BNI1, IQG1, and HOF1) associated with the regulation of cytokinesis were identified in the evolved C. auris aggregative strains (Fig. 3). Lrg1 is a GTPase-activating protein (GAP) that regulates Cdc42 and Ras1, which control morphogenesis through multiple signaling pathways in fungi45,46. The formin protein Bni1, IQGAP protein Iqg1, and F-BAR protein Hof1 are downstream of Lrg1. Lrg1 and Bni1 regulate cellular polarity, while Hof1 is required for assembly and contraction of the actomyosin ring (AMR)45,46,47. We note that several genes identified in the mutated list are involved in the regulation of multiple biological processes, such as LRG1 and BNI1 (Fig. 3b, c).
In addition to LRG1 and BNI1, mutations in two other genes (HSL1 and KIN3) were also identified in the in vivo evolved strains. Hsl1 and Kin3 play critical roles in cellular polarity and budding in several yeast species48. Bni1 are core components of the polarisome49, while Hsl1 and Kin3 are kinases involved in the regulation of septin assembly and filamentation50,51. Mutations in seven genes Pan1, Arc19 and AP adaptors (Apl2, Apl4, Laa1, Apm1, and Bsp1), which are core proteins involved in cytoskeletal organization, were also identified (Fig. 3). Moreover, we identified 10 genes involved in the regulation of cell wall integrity, including DIG2 (encoding a MAPK proteins), LRG1, BNI1 and several genes associated with cell wall formation (CHS1, MNN11, KRE5, KTR2, OCH1, VAN1, and VRG4). Of these genes, three (OCH1, MNN11, and VAN1) are required for the initiation and elongation of N-mannosylation of cell wall in yeasts52. Gene ontology category analysis further confirmed the genetic and functional associations we observed among the identified mutated genes (Fig. S2). Moreover, we found that evolved aggregative mutants of the same genes that derived from different parental strains (of different genetic clades) exhibited a similar phenotype (Fig. S3), indicating the general characteristic of this phenomenon in clinical strains.
A mutation in SSD1 (p.R549K) was found in the aggregative cells of the C. auris clinical isolate AR0386A. SSD1 encodes a translational repressor and is involved in polar growth in S. cerevisiae53,54. This mutation was located within a region of SSD1 that encodes the conserved domain III of the protein, which could attenuate or inactivate Ssd1 function and thus lead to the observed aggregative phenotype (Fig. 1). A similar aggregative phenotype was observed in the evolved strain carrying the same site mutation we generated in the control strain (Fig. S4). These findings demonstrate that the p.R549K mutation in SSD1 is directly related to the aggregative phenotype in C. auris.
To reveal whether mutations in cell budding- or cell division-associated genes are prevalent in C. auris clinical strains, we next analyzed 4482 genomic sequences of C. auris available in the NCBI database. As shown in Fig. S5 and Dataset S2, based on the mutated genes identified in our evolved aggregative strains, we identified 11 strains containing gene mutations associated with cell budding or cell division processes. Specifically, 9 types of mutations (ACE2 Q163*, BNI1 p.T484 fs, CAS4 p.E1289 fs, CAS4 p.Q2657*, ENO1 p.K270 fs, INP52 p.E921*, LRG1 p.V434 fs, LRG1 p.S296 fs, PAN1 p.E1047 fs) were identified (only nonsense and frameshift mutations were considered), several of which overlapped with those identified in the evolved aggregative strains (Fig. 3). These findings indicate that mutations in cell budding- or cell division-associated regulators are present in C. auris clinical strains.
Taken together, most mutations identified in the C. auris evolved aggregative isolates likely impact biological processes involved in regulating cell budding or cell division, such as cytokinesis, cytoskeletal properties, cellular polarity, and cell wall integrity (Fig. 3b, c). Most of the genes identified in our mutational screens are conserved in C. albicans and S. cerevisiae and play critical roles in the regulation of cell aggregation, and filamentous or invasive growth in these yeast species (Table S3). It is reasonable to predict that genetic perturbations of these processes would lead to the evolution of the multicellular aggregative morphology, which could benefit C. auris by allowing it to adapt to the changing host microenvironment and thus persist and survive in the mammalian host.
Verification of the association between mutated genes and the aggregative phenotype of the evolved strains
To test whether inactivation of the aforementioned major genes identified in the evolved strains is sufficient to cause the aggregative phenotype, we constructed a set of deletion or substitution mutant strains of 14 representative genes (ace2Δ, apm1Δ, apl2Δ, bni1Δ, cas4Δ, chs1Δ, hof1Δ, iqg1Δ, irg1Δ, och1Δ, nip100Δ, sgs1Δ, ssd1Δ, and van1Δ) in a his1 deletion strain of BJCA001. These genes play central roles in the regulation of cytokinesis, cytoskeletal properties, cellular polarity, and/or cell wall integrity in fungal species. As shown in Fig. S4, all mutant strains constructed showed similar cellular and colony morphologies to those of their corresponding evolved aggregative strain counterparts. The mutant strains also exhibited cell budding and/or cell division defects, suggesting that the identified host-induced genetic mutations of the evolved strains were sufficient to cause the generation of the aggregative morphology in C. auris.
Global transcriptional expression profiles of the yeast-form and aggregative isolates of C. auris
To further explore the mechanism of formation of the aggregative phenotype, we performed transcriptomic analysis using RNA-Seq assays in the yeast-form strain (BJCA001) and four aggregative isolates (CHS1 p.Q826*, BNI1 p.206 fs, LRG1 p.K427*, and CAS4 p.1288 fs mutants). Three biological repeats were performed. Principal Component Analysis (PCA) analysis indicated the consistency of expression profiles among the repeats (Fig. S6). As shown in supplementary Dataset S3, 62, 649, 833, and 1994 differentially expressed genes (DEGs) were identified between the WT (yeast-form) and CHS1 p.Q826*, WT and BNI1 p.206 fs, WT and LRG1 p.K427*, and WT and CAS4 p.1288 fs mutants, respectively. Hierarchical clustering analysis of DEGs indicated a high transcriptional expression similarity between the yeast-form strain and CHS1 p.Q826* mutant and also between the BNI1 p.206 fs and LRG1 p.K427* mutants (Fig. 4a). There were 445 common DEGs between the BNI1 p.206 fs and LRG1 p.K427* mutants (Fig. 4b, c). Many of them are involved in the regulation of cytoskeleton, cell cycle or cytokinesis, and cell wall integrity (CWI) (Fig. 4b and Dataset S3). These results are reasonable because Bni1 and Lrg1 are associated regulators involved in the regulation of the CWI55, cytokinesis, and polarity pathways, while Cas4 is a member of the RAM pathway that regulates cell polarity and separation42. Mutation of CAS4 affected the expression of nearly 2000 genes perhaps due to its global regulatory role in C. albicans. Mutation of CHS1 only caused 62 DEGs perhaps due to the chitin synthase Chs1 representing a downstream component of the CWI pathway56. Consistent with the aggregative phenotype, we found a set of common DEGs associated with cytoskeleton, cell division, and cell wall integrity in all the four evolved mutants (compared to that in the yeast-form strain BJCA001, Fig. 4d and Dataset S3). Of them, CDC5, CDC14, CDC20, ESC4, and HGC1 are involved in the regulation of cell division. HGC1 encodes a G1 cyclin-related protein that is associated with the cyclin-dependent kinase Cdc28 and is essential for filamentous growth in C. albicans15.

Strains analyzed: BJCA001 (yeast-form), CHS1 p.Q826* (chs1, FDAG4), BNI1 p.206 fs (bni1, FDAG30), LRG1 p.K427* (lrg1, FDAG9), and CAS4 p.1288 fs (cas4, FDAG1). a Hierarchical clustering of differentially expressed genes (DEGs) between different strains using Euclidean distance. b Numbers of DEGs associated with CWI, cytoskeleton, and cell cycle/cytokinesis regulation between the yeast-form strain and CHS1 p.Q826*, BNI1 p.206 fs, LRG1 p.K427*, or CAS4 p.1288 fs mutants. c Venn plot of DEGs between the yeast-form strain and BNI1 p.206 fs, LRG1 p.K427*, or CAS4 p.1288 fs mutants. d Representative common DEGs among the CHS1 p.Q826*, BNI1 p.206 fs, LRG1 p.K427*, and CAS4 p.1288 fs mutants. e Representative common DEGs among the BNI1 p.206 fs, LRG1 p.K427*, and CAS4 p.1288 fs mutants. f Representative common DEGs between the LRG1 p.K427* and CAS4 p.1288 fs mutants. Log2 (fold change) values of DEGs between the yeast-form strain and corresponding mutant strains are shown in (d)–(f).
Compared to the yeast-form strain, 398 common DEGs were identified in the BNI1 p.206 fs, LRG1 p.K427*, and CAS4 p.1288 fs mutants (Fig. 4e). Of these DEGs, a large subset of genes are associated with the regulation of cell wall integrity (e.g., PKC1, BMT1, CHS1, and PGA6) and some are regulators of cell budding (such as IQG1 and GIN4). Moreover, many DEGs are associated basic metabolic processes (such as amino acid metabolism and protein synthesis), suggesting that mutation of these genes has a great impact on cell physiology. A large set of genes involved in the regulation of cell wall integrity, cell cycle, and cytoskeleton (e.g., IQG1, OCH1, CHS1, LMO1, ENO1, CAS4, CHS3, and CHS6) exhibited a similar expression pattern in the BNI1 p.206 fs, LRG1 p.K427*, and CAS4 p.1288 fs mutants (Fig. 4e, f). In contrast, mutation of CHS1 had a relatively weak impact on the general cell physiology. To further uncover the genetic and functional connections among the DEGs between the yeast-form strain and aggregative mutants (common DEGs found in two or more mutants), we performed network analysis using the STRING database57. Three major clusters involving 96 proteins with predicted protein-protein interaction relationship were identified by K-mean method. As shown in Fig. S7, cluster 1 consisted of 7 genes regulating the aggregative phenotype identified in the evolved strains in this study and some key regulators of cytokinesis and the CWI pathway. Cluster 2 proteins were mainly associated with cell cycle and cytokinesis. Cluster 3 proteins were majorly related to the cell wall integrity. These findings confirmed that the mutated genes of the aggregative isolates play critical roles in the regulation of CWI, cytoskeleton, cytokinesis, and cell polarity. Genetic perturbation of these pathways would lead to cell budding or division defects and formation of multicellular aggregates.
Aggregative cells display an increased fitness and outcompete single-celled yeast-form cells in the brain during system infection
The ability to undergo morphological transitions is a common strategy used by pathogenic fungi to survive in the host and evade host immune attacks11,58,59. To compare the fitness and virulence of aggregative and single-celled yeast-form cells of C. auris, we performed systemic infection and fungal burden assays using a mouse infection model using a selection of WT yeast-form and evolved mutant aggregative strains. To avoid blood vessel blockage and rapid animal death caused by C. auris aggregates, we converted C. auris aggregative cells to single cells by sonication prior to tail veil injection in the mice (Fig. 5a). C. auris cells were recovered from the brain, liver, spleen, lung, and kidney, and fungal burdens were calculated at 1, 3, and 7 days post infection (dpi).

a Schematic of the systemic infection assay. To convert to separated single cells for systemic infections, C. auris aggregative cells were subjected to sonication. To control for any potential negative effects of sonication, yeast-form cells were also treated with the same sonication protocol. Fungal cells (1 × 107) were injected into each mouse via the tail vein. At 1, 3, or 7 dpi, fungal cells were recovered from the brain, liver, spleen, lung, and kidney tissues, weighed, and pulverized using Zirconia beads (3.2 mm) with 60 Hz power for 120 s. The homogenized samples were then re-plated onto...
Popular posts from this blog
Mysterious Morel Mushrooms at Center of Food Poisoning Outbreak - Kaiser Health News

Comments
Post a Comment